Амилоидоз — групповое понятие, объединяющее заболевания, отличительным признаком которых является внеклеточное отложение в тканях специфического нерастворимого фибриллярного белка — амилоида. Почки часто вовлекаются при генерализованных формах амилоидоза; амилоидная нефропатия может превалировать в клинической картине или конкурировать с поражением других жизненно важных органов — сердца, нервной системы, желудочно-кишечного тракта, значительно утяжеляя течение заболевания и определяя прогноз.
Термин «амилоид» ввел R. Virchow, который выявил свойство вещества, выпадающего в тканях при так называемой сальной болезни (у больных туберкулезом, актиномикозом, сифилисом), окрашиваться йодом, подобно крахмалу. Несмотря на то что в дальнейшем была выяснена белковая природа амилоида, термин закрепился в медицинской литературе. Особенностью амилоидной фибриллы по сравнению с другими фибриллярными белками интерстиция является своеобразная пространственная организация молекулы, проявляющаяся в ее кросс-в-складчатой конформации. Благодаря в-складчатой структуре фибрилла амилоида обладает свойством двойного лучепреломления, т.е. расщепляет падающий световой луч на два, один из которых поглощается молекулой (дихроизм молекулы). Это свойство используется в диагностике амилоидоза: в поляризованном свете наличие амилоида в морфологических препаратах, окрашенных красным конго, определяется по характерному зеленому свечению.
Распределение амилоидоза среди населения земного шара изучено недостаточно.
В США заболеваемость AL-амилоидозом составляет 5,1—12,8 на 1 млн населения в год (в среднем 1275—3200 новых случаев ежегодно), частота АА-амилоидоза оценивается в 9—11 раз выше. По данным ЕДТА, среди причин почечной недостаточности в 1992 г. на амилоидоз почек пришлось около 1 %, в некоторых странах Северной Европы (Финляндия, Норвегия, Швеция) — значительно выше (в Финляндии около 19 %) за счет распространенного в этих странах семейного ATTR-амилоидоза.
Согласно регистру Медицинского научного совета Великобритании по гломерулонефриту, в 1985 г. амилоидоз почек выявлен у 2,8 % среди 3362 больных, кому проведены биопсии почек. По данным регистра биопсий почек, в Италии частота амилоидоза почек составила в среднем 2,5 % (от 3,2 до 2,2 %) среди 14 477 биопсий, выполненных за период 1987—1993 гг. в 71 центре страны. По материалам прозектуры MMA им. И.М. Сеченова за 20 лет, на долю амилоидоза пришлось 1,08 % всех вскрытий, из них на первичный AL-амилоидоз — 0,1 %.
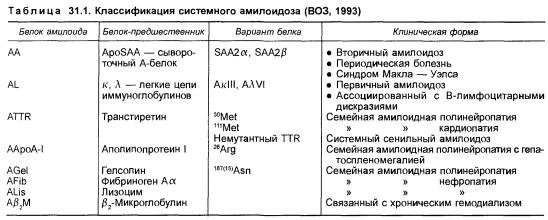
Классификация амилоидоза, этиология, патогенез. Амилоидная фибрилла представляет собой продукт конденсации различных белков. Основной фибриллярный белок, содержание которого в амилоиде достигает 80 %, образуется из белка-предшественника, специфичного для каждого типа амилоида.
Строгое соответствие основного белка-предшественника определенному клинико-морфологическому типу амилоида дает ключ к пониманию патогенеза разных форм амилоидоза. На принципе дифференциации типов амилоида и соответствующих белков-предшественников построены современные классификации амилоидоза.
В классификации ВОЗ 1993 г. каждый тип амилоида имеет буквенное обозначение, в котором первая прописная буква А указывает на «амилоид», последующие — на краткое название основного фибриллярного белка амилоида. Выделяют системные (генерализованные) и локальные формы амилоидоза.
К системному (генерализованному) амилоидозу относятся (табл. 31.1) АА-амилоидоз, AL-амилоидоз, семейный ATTR-амилоидоз и другие формы семейного амилоидоза (распространенные в определенных районах земного шара — в Швеции, Дании, Португалии, Финляндии и др.), сенильный ATTR-амилоидоз, в2М-амилоидоз (диализный).

АА-амилоидоз объединяет вторичный реактивный амилоидоз, амилоидоз в рамках периодической болезни и синдрома Макла — Уэлса. Среди причин вторичного АА-амилоидоза большая роль принадлежит ревматоидному артриту (PA), анкилозирующему спондилоартриту (болезнь Бехтерева), псориатическому артриту и опухолям, в том числе гематологическим (лимфома, лимфогранулематоз). В странах Европы амилоидоз развивается у 10 % больных PA и ювенильным PA (ЮРА). По материалам клиники им. Е.М. Тареева MMA им. И.М. Сеченова, PA, ЮРА и болезнь Бехтерева занимают первое место в ряду предрасполагающих к вторичному АА-амилоидозу заболеваний, составляя 43 % среди 146 больных, паранеопластический АА-амилоидоз — 17 % (из 25 больных с опухолями у 14 был выявлен лимфогранулематоз, у 7 — опухоль почек, у 2 — менингиома, у 1 — хемодектома, у 1 больного — хронический лимфолейкоз). Возрастающее значение в структуре причин АА-амилоидоза приобретают хронические заболевания кишечника — неспецифический язвенный колит и болезнь Крона. Сохраняется значение хронических гнойно-деструктивных процессов и туберкулеза, хотя они отступают на второй план в связи с внедрением эффективных методов лечения.
АА-амилоидоз развивается у 40 % больных периодической болезнью, средиземноморской лихорадкой, которая наблюдается у выходцев из района Средиземного моря, у нас в стране — среди армян и евреев, имеет рецессивный тип наследования и характеризуется рецидивирующими приступами лихорадки, болей в животе, грудной клетке, суставах. К АА-амилоидозу относится также синдром Макла — Уэлса — доминантно-наследуемая семейная нефропатия с крапивницей и глухотой. В России 2 случая семейного амилоидоза, клинически сходного с синдромом Макла — Уэлса, были описаны О.М. Виноградовой.
АА-амилоид образуется из сывороточного предшественника (SAA) — острофазового белка, продуцируемого в ответ на воспаление, при котором его количество повышается во много раз; SAA является а-глобулином, по своим функциональным свойствам близким С-реактивному белку; у человека и животных синтезируется клетками разных типов (гепатоцитами, нейтрофилами, фибробластами).
Органами-мишенями АА-амилоидоза являются почки, печень, селезенка, кишечник, надпочечники.
AL-амилоидоз включает первичный (идиопатический) амилоидоз, амилоидоз при миеломной болезни и В-клеточных опухолях (болезнь Вальденстрема и др.). Большинство исследователей рассматривают AL-амилоидоз (и первичный, и связанный с миеломой) в рамках единой В-лимфоцитарной дискразии. Полагают, что аномальный клон плазматических или B-клеток в костном мозге продуцирует иммуноглобулины, обладающие амилоидогенностью. Предшественниками AL-амилоида служат вариабельные участки легких цепей моноклонального иммуноглобулина, чаще A-, реже к-типов.
Основными органами-мишенями при AL-амилоидозе являются сердце, язык, желудочно-кишечный тракт, нервная система, кожа, а также почки.
К АТТR-амилоидозу относятся семейная амилоидная полинейропатия (реже кардиопатия и нефропатия) с аутосомно-доминантным типом наследования, системный старческий амилоидоз. Сывороточным белком-предшественником амилоидоза в этой группе является компонент молекулы преальбумина транстиретин — транспортный белок для тироксина и ретинола, первично синтезируемый в печени. Установлено, что наследственный семейный амилоидоз является результатом мутации в гене, ответственном за синтез молекулы транстиретина. Мутантные формы и других белков — аполипопротеина А-1, гелсолина, фибриногена Aa и лизоцима также могут вести к развитию наследственного амилоидоза, в том числе нефропатического (см. табл. 31.1), однако мутантный (вариантный) TTR является наиболее частым предшественником семейного амилоидоза.
В основе системного старческого амилоидоза лежит немутантный (невариантный) транстиретин. Старческий системный амилоидоз имеет в качестве основных органов-мишеней сердце, сосуды, отличается от кардиопатии при AL-амилоидозе в целом меньшей тяжестью поражения, нередким сочетанием с атеросклерозом.
Ав2М(диализный)-амилоидоз — форма системного амилоидоза, развивающаяся у пациентов, находящихся на длительном гемодиализе (в среднем через 7 лет). Белком-предшественником является в2-микроглобулин (в2М), который плохо фильтруется через большинство диализных мембран современного типа и задерживается в организме. Основными органами-мишенями являются кости, периартикулярньге ткани (могут возникать переломы костей, требующие ортопедических мероприятий вплоть до протезирования). Часто наблюдается синдром карпального канала (онемение и боль в первых 3 пальцах кисти, распространяющиеся на предплечье, с последующим развитием атрофии мышц тенара) из-за сдавления срединного нерва отложениями амилоидных масс в области карпальной связки.
Амилоидогенез является сложным и многоступенчатым процессом, многие звенья которого остаются неясными.
Известные в настоящее время белки-предшественники системных форм амилоидоза, циркулирующие в крови больных амилоидозом, обнаруживаются и в крови здоровых индивидов. Среди причин появления (усиления) амилоидогенности белков-предшественников крови в настоящее время рассматривают их молекулярную гетерогенность, т.е. существование амилоидогенных аллотипов белка. Амилоидогенный вариант белка-предшественника может наследоваться либо возникать в результате спонтанных мутаций в течение жизни.
Так, в эксперименте показано, что L-цепи подтипов AVI и кI вследствие отдельных аминокислотных замен в вариабельных участках отличаются молекулярной нестабильностью и способны образовывать фибриллярные белковые комплексы in vitro. Именно эти подтипы L-цепей чаще циркулируют в крови больных идиопатическим и ассоциированным с В-клеточными опухолями амилоидозом. Большинство семейных форм амилоидоза характеризуется циркуляцией в крови вариантных форм транстиретина с измененной аминокислотной последовательностью. На примере этой формы амилоидоза было показано, что нарастание амилоидогенности мутантного транстиретина может быть связано с нарушением поверхностного pH, появлением аномальных электрических зарядов в активных центрах белковых субъединиц, изменением гидрофильности молекулы. В результате дестабилизируется архитектоника белковой молекулы, повышается ее химическая реактогенность, что способствует конденсации образующихся продуктов в малорастворимые макромолекулярные белковые комплексы с образованием в конечном итоге амилоидных фибрилл.
Известно более 50 вариантов транстиретина с аминокислотной заменой в том или ином положении. Биохимическая вариабельность транстиретина влияет на характер клинических проявлений семейного амилоидоза. Так, при португальском типе, проявляющемся полинейропатией с преимущественным поражением нервных стволов нижних конечностей и органов таза, белком-предшественником является транстиретин с заменой метионина на валин в 30-м положении (ATTR [30Met]-амилоидоз), в то же время преимущественно карди-опатический датский тип амилоидоза ассоциируется с транстиретином, в котором метионин в 111-м положении заменен на лейцин (ATTR[IIIMet]-амилоидоз).
Считается, что старческий системный амилоидоз ассоциируется с транстиретином, имеющим неизмененную аминокислотную последовательность, однако недавно у пожилых американцев негроидной расы больных ATTR-амилоидозом был выявлен амилоид, построенный из остатков транстиретина с заменой изолейцина в 122-м положении на валин. Показано, что носителями этого аллеля гена, ответственного за синтез транстиретина, являются 3,9 % американцев черной расы, но обусловленная им амилоидная кардиопатия из-за позднего начала распознается редко, так как маскируется атеросклерозом. Таким образом, патогенетические различия между старческим системным амилоидозом и семейным ATTR-амилоидозом стираются, в связи с чем эти формы амилоидоза все больше считают вариантами одной болезни.
В последние годы было показано, что популяция белка SAA также гетерогенна, как это установлено для L-цепей и транстиретина. Фрагменты не менее 5 молекулярных форм SAA обнаруживают в амилоидных депозитах. Возможно, что именно эти молекулярные формы SAA амилоидогенны.
Конечный этап амилоидогенеза — образование фибрилл амилоида клетками-амилоидобластами в межклеточном матриксе. Так, считают, что образование AA из SAA осуществляется путем неполного расщепления протеазами, связанными с поверхностной мембраной моноцитов-макрофагов. Полимеризация растворимого АА-белка в фибриллы происходит также на поверхности макрофагов механизмом перекрестного связывания полипептидов при участии мембранных ферментов. В эксперименте с казеиновым амилоидозом у мышей показана важная роль в индукции АА-отложений так называемого амилоидускоряющего фактора (АУФ), образующегося в селезенке и печени.
Высказывается мнение, что в2М приобретает амилоидогенные свойства вследствие частичного протеолиза, обусловленного возможным действием цитокинов (фактор некроза опухоли а, интерлейкин-1, интерлейкин-6 и др.), высвобождение которых стимулируется различными субстратами — диализной мембраной, компонентами диализной жидкости и др. Показано, что в2M проявляет значительную коллагенсвязывающую активность, чем может быть обусловлено преимущественное отложение амилоида в костно-суставных структурах.
Семейный ATTR-амилоидоз, несмотря на его наследственную природу, обычно проявляется только к середине жизни; развитие системного старческого ATTR-амилоидоза наблюдается исключительно у пожилых людей старше 70 лет, что рассматривают как убедительный аргумент в пользу существования «возрастных» триггеров амилоидогенеза. В амилоидогенезе при старческом амилоидозе, по-видимому, имеют значение конформационные нарушения белков при нарастающей с возрастом брадитрофии тканей.
Почки вовлекаются в разной степени при всех формах генерализованного амилоидоза, однако наибольшее клиническое значение имеют AA- и AL-амилоидоз почек. Амилоидная нефропатия — основное проявление АА-амилоидоза, но и при AL-амилоидозе, который традиционно рассматривают как кардиопатический амилоидоз, вовлечение почек наблюдается примерно с той же частотой, что и сердца [Виноградова О.М., 1980]. Более того, в наблюдении G. Banfi, G. Marinone среди 40 пациентов с AL-амилоидозом у 30 % почки были единственным органом, поражение которого клинически выявлялось на момент постановки диагноза. При вскрытии амилоидные отложения в почках находят в 80 % случаев первичного AL-амилоидоза.
Морфологическая картина. В большинстве случаев амилоидоза почки увеличены, выглядят светлыми с гладкой ровной поверхностью, общая почечная масса может достигать 600 г. Граница между корой и медуллярным слоем плохо различима, размыта. Иногда (в 10 % случаев) почки имеют неровную поверхность из-за очаговой атрофии коры, связанной с артериолосклерозом и/или окклюзией почечных сосудов амилоидными массами. Нередко выявляется тромбоз почечных вен.
При AA- и AL-амилоидозе амилоид может располагаться во всех отделах почек, но преимущественно поражаются клубочки. При AL-амилоидозе амилоидные отложения могут ограничиваться внегломерулярными областями, не затрагивая самих клубочков.
В световом микроскопе амилоид выглядит как аморфные слегка эозинофильные массы, принадлежность которых к амилоиду подтверждается при окраске конго красным (розовокрасное окрашивание) с последующей микроскопией в поляризованном свете (появление зеленого свечения из-за характерного свойства двойного лучепреломления). Другие методы выявления амилоида — с помощью тиофлавина-Т (коричнево-зеленое окрашивание), кристаллического фиолетового (коричнево-красное окрашивание) — по специфичности уступают окраске конго красным. Типировать AL- и AA-амилоид возможно, применяя окраску конго красным с добавлением трипсина или перманганата калия, при этом АА-амилоид утрачивает свойство двойного лучепреломления и теряет окраску, в то время как AL-амилоид благодаря устойчивости к протеолизу сохраняет окраску. Сходный принцип используется и в методе дифференциации AL- и АА-амилоидоза с добавлением щелочного гуанидина. Эффективным методом является применение специфических антисывороток (антитела к АА-амилоиду и легким цепям иммуноглобулинов) в комбинации с иммунопероксидазной окраской, однако и с помощью этого подхода не всегда удается идентифицировать AL-тип амилоида.
На ранней стадии поражения почек отложения амилоида обнаруживаются в мезангии в виде очагов (узлов) вблизи основания сосудистого пучка. При этом отсутствует пролиферация мезангиальных клеток, базальная мембрана клубочка (БМК) интактна. По мере прогрессирования происходит распространение амилоида через капиллярный пучок к периферии клубочка. Вначале наблюдается очаговая инфильтрация капиллярной стенки с расположением амилоида вдоль эндотелиальной поверхности БМК, затем отдельные массы амилоида появляются в субэпителиальном пространстве, вблизи больших отростков подоцитов, постепенно они охватывают всю длину стенки. На более поздней стадии нормальная структура клубочка стирается, амилоидные массы сливаются с БМК. Иногда на границе с амилоидными депозитами видны гигантские многоядерные клетки, участвующие в резорбции амилоидных фибрилл амилоида. Могут выявляться (редко) и клеточные полулуния в капсуле клубочка (боуменовой капсуле). В конечном итоге развивается полная облитерация клубочков, причем они остаются увеличенными, на периферии формируются зоны коллагена. Как правило, амилоидом инфильтрированы сосуды (артерии и вены) разных калибров, при этом с самого начала поражается вся толща сосудистой стенки, включая адвентиций.
Отложения амилоида обнаруживаются и в других структурах почки — вдоль тубулярной базальной мембраны дистальных канальцев и петли Генле с возможной атрофией канальцев, интерстициальной ткани.
В электронном микроскопе фибриллы амилоида имеют 8—10 пм в ширину и 30 пм (до 1 мкм) в длину, отличаются компактным и параллельным расположением; при световой микроскопии они могут напоминать «шиповидные» образования при мембранозной нефропатии.
Показано, что проникновение амилоида в субэпителиальное пространство вызывает слияние ножек подоцитов, конденсацию в них микрофиламентов, отделение от lamina гага externa с фокальным обнажением БМК. Эти изменения коррелируют с выраженностью протеинурии. Подоциты, по-видимому, играют основную роль и в процессах репарации в клубочках. Во время репаративной фазы, которая продолжается в течение нескольких лет, подоциты медленно восстанавливаются, начинают синтезировать новую БМК, которая образует второй мембранный слой, что приводит к снижению протеинурии и улучшению почечной функции.
Клиническая картина. Амилоидоз почек манифестирует обычно протеинурией, которая при АА-амилоидозе обнаруживается чаще и более выражена, чем при AL-амилоидозе. Так, частота выявления протеинурии при АА-типе составляет 100 %, при AL-амилоидозе — 82 %. Протеинурия как наиболее достоверный признак поражения почек выявляется в разные сроки: по нашим данным, одинаково часто как в первые 3 года, так и после 10 лет существования основного заболевания. В ранний период болезни протеинурия может эпизодически уменьшаться или усиливаться, особенно при обострении основного заболевания. Ho обычно она довольно скоро принимает постоянный и достаточно выраженный характер. Значительная потеря белка почками, а также ряд других факторов (усиление катаболизма белка в организме, уменьшение всасывания и усиленное выделение белка через желудочно-кишечный тракт) приводят к развитию гипопротеинемии с гипоальбуминемией (до 10 г/л) и связанного с ними отечного синдрома. Более характерно постепенное развитие нефротического синдрома (НС) вслед за стадией умеренной протеинурии, иногда весьма длительной.
Ho достаточно часто — у 46—50 % больных с АА-амилоидозом и у 32 % — с AL-амилоидозом — заболевание диагностируется на стадии НС. По сводным данным F.P.Schena и соавт. (1996), среди 259 пациентов с почечным амилоидозом (и AA-, и AL-типов) HC отмечен у 80 %. Возможная связь нефротического синдрома с амилоидозом увеличивается с возрастом. Так, среди 317 пациентов пожилого возраста (старше 60 лет) амилоидоз как причина развития у них HC выявлен у 10,7 %. НС, обусловленный амилоидозом, может протекать со всеми классическими симптомами — массивной протеинурией, гипоальбуминемией, гиперхолестеринемией и большими отеками. Как правило, отеки развиваются довольно рано, приобретают распространенный и упорный характер, оставаясь значительными и в терминальном уремическом периоде. Хотя отеки при амилоидозе возникают у большинства больных, их отсутствие возможно в связи с потерей натрия (инфильтрация амилоидом надпочечников).
Среди других манифестаций амилоидоза почек наблюдается почечная недостаточность. По данным F.P. Schena и соавт., ХПН к моменту подтверждения амилоидоза почек выявлялась у 33 % и ОПН — у 6 % больных.
В 12,5 % случаев возможно начало клинических проявлений амилоидоза почек с почечной недостаточностью без протеинурии, что объясняют преимущественным поражением амилоидом сосудов без существенного вовлечения клубочков, чаще при AL-амилоидозе. Иногда при амилоидозе почек независимо от его типа отмечается быстрое прогрессирование почечной недостаточности в результате тромбоза почечных вен (реже) и/или выраженной атрофии канальцев (чаще). Тубу-лоинтерстициальные нарушения с развитием интерстициального фиброза в большей степени, чем количество отложений амилоида в клубочках, коррелируют с выраженностью почечной недостаточности, поэтому выявление канальцевой дисфункции, в том числе и с помощью радионуклидных методов, имеет важное прогностическое значение.
Несмотря на хроническую почечную недостаточность, размеры почек при УЗИ остаются увеличенными в большинстве случаев. К особенностям амилоидной нефропатии, как уже было сказано, относится также сохранение нефротического синдрома при развитии почечной недостаточности, даже если уровень клубочковой фильтрации снижается до критического.
Для амилоидоза почек характерен скудный мочевой осадок, в том числе у больных с массивной протеинурией, однако это классическое представление правомерно не всегда. Может выявляться (по разным данным, в 3,3—11,5 % случаев) стойкая микрогематурия, иногда (у 0,3 % больных) макрогематурия, которая требует исключения опухолевого процесса. Асептическая лейкоцитурия встречается у 30—40 % больных без сопутствующего пиелонефрита. Соответственно степени протеинурии обнаруживаются гиалиновые и реже зернистые цилиндры, дающие резко положительную ШИК-реакцию, а также липидурия с наличием двоякопреломляющих кристаллов и капель в осадке мочи.
Примерно у 25 % больных с AL-амилоидозом отмечается ортостатическая гипотония вследствие нарушений вегетативной нервной системы (диффузная автономная нейропатия). Даже при прогрессировании почечной недостаточности со значительным повышением уровня сывороточного креатинина системная гипертония у больных с AL-амилоидозом развивается редко. У больных с АА-амилоидозом артериальная гипертония наблюдается в 23 % случаев, чаще в конечной стадии болезни как проявление ХПН, но может быть обнаружена у 15 % еще до появления ХПН. Обычно артериальная гипертония невысокая, хотя может быть и злокачественной; редки, но возможны, тяжелые гипертонические кризы.
Внепочечные проявления амилоидоза зависят от типа амилоида и стадии болезни. Для системного амилоидоза, особенно первичного, характерно последовательное присоединение новых симптомов, создающих полиморфную клиническую картину с многоорганными проявлениями, что важно иметь в виду при диагностике амилоидоза, в том числе с преимущественным поражением почек.
Сердце часто вовлекается в процесс при AL-амилоидозе; при АА-типе клинически значимое поражение сердца наблюдается редко. Амилоидоз сердца при AL-амилоидозе характеризуется в первую очередь развитием рефрактерной к лечению застойной сердечной недостаточности по рестриктивному типу. У 23 % больных сердечная недостаточность диагностируется уже в дебюте болезни и затем быстро прогрессирует, являясь причиной смерти 40 % пациентов. В зависимости от локализации депозитов амилоида в миокарде могут наблюдаться синдром слабости синусового узла, атриовентрикулярная блокада, разнообразные аритмии, очаговые поражения миокарда (псевдоинфаркт).
На ЭКГ наиболее часто встречаются снижение вольтажа зубцов, нарушения ритма и проводимости. При ЭхоКГ отмечается картина рестриктивной кардиопатии с признаками диастолической дисфункции, которая лучше диагностируется по изменению трансмитрального кровотока при допплерэхокардиографии.
Периферическая и вегетативная нервная система может поражаться при AL-амилоидозе: отмечаются повреждения аксонов с поражением тонких миелинизированных и немиелинизированных нервных стволов. Периферическая полинейропатия — сенсорная, редко моторная (как правило, симметричная, начинающаяся с дистальных отделов конечностей и распространяющаяся на проксимальные), развивается у 17 % больных и может преобладать в клинической картине, создавая диагностические трудности. Нарушения вегетативной нервной системы (автономные дисфункции) проявляются симптомами ортостатической гипотонии, импотенцией, сфинктерными расстройствами. У 20 % больных AL-амилоидозом развивается синдром карпального канала (парестезии и боли в кистях по ходу ветвей срединного нерва), который может предшествовать развитию других симптомов болезни; при АА-амилоидозе он не наблюдается.
Желудочно-кишечный тракт при амилоидозе поражается на всем протяжении. Макроглоссия — нечастый (у 22 %), но очень характерный признак AL-амилоидоза, при АА-амилоидозе макроглоссия не развивается. Может поражаться пищевод, иногда находят опухолевидное отложение амилоида в желудке или кишечнике. Возникновение поноса и других проявлений кишечной диспепсии у больных амилоидозом обусловлено гипермоторной энтеропатией вследствие отложения амилоида по ходу нервных сплетений кишечника. Диарея как проявление изолированного синдрома нарушенного всасывания в тонком кишечнике встречается лишь у 5 % больных AL-амилоидозом и у 13—20 % больных АА-амилоидозом, так как отложения амилоида в ворсинках тонкого кишечника редки, депозиты амилоида чаще обнаруживаются в подслизистом слое. В связи с этим для выявления амилоида рекомендуют проводить биопсию слизистой оболочки кишечника с подслизистым слоем.
Поражение амилоидом поперечнополосатой мускулатуры характерно для AL-амилоидоза. Мышцы больных выглядят плотными, увеличенными, отмечается выраженная мышечная слабость (псевдогипертрофия).
Печень закономерно вовлекается в патологический процесс у 30 % больных AL- и у 50— 60 % АА-амилоидозом. Гепатомегалия появляется уже на ранних стадиях амилоидоза, однако белково-синтетическая и другие функции печени на протяжение всего заболевания, как правило, остаются сохранными. Амилоидоз печени чаще проявляется симптомами умеренного холестаза.
Селезенка увеличена только у 5 % больных AL-амилоидозом, у больных АА-амилоидозом спленомегалия выявляется у 30—40 %.
Респираторный тракт чаще вовлекается в процесс при AL-амилоидозе — у 50 % больных (при АА-амилоидозе — у 10—14 %). Охриплость голоса, связанная с вовлечением голосовых связок, по данным больших статистик, опережает проявления, обусловленные отложениями амилоида в дистальных отделах — бронхах, альвеолярных перегородках и сосудах. Возможен опухолевидный легочный амилоидоз, имитирующий картину рака легкого.
У больных AL-амилоидозом наблюдаются развитие геморрагического синдрома с характерными кровоизлияниями вокруг глаз («глаза енота», «симптом очков»), вовлечение щитовидной железы, надпочечников (с их недостаточностью), самые различные изменения кожи (папулы, узлы, бляшки, диффузная инфильтрация кожи с трофическими изменениями), суставов.
Изменения лабораторных показателей неспецифичны: увеличение СОЭ, гиперглобулинемия, тромбоцитоз, который наряду с малыми размерами тромбоцитов и появлением в циркуляции эритроцитов с тельцами Жолли рассматривают как свидетельство функционального гипоспленизма, дефицит X фактора свертывания при AL-амилоидозе (как и поражение сосудов, его считают причиной развития геморрагий). У больных с AL-амилоидозом выявляется плазматизация костного мозга. При первичном амилоидозе плазматические клетки в миелограмме составляют в среднем 5 %, у 1/4 больных они превышают 10 % (11—22 %), но при этом не отмечается лизиса костей и столь высокого, как при миеломной болезни, уровня моноклонального иммуноглобулина в сыворотке крови (выше 3 г/дл) и моче (более 2,5 г/сут). При иммуноэлектрофорезе концентрированной мочи моноклональные легкие цепи (белок Бенс-Джонса) выявляются у 72 % больных AL-амилоидозом — у 93 % больных с миеломой соотношение к- и А-цепей составило 50 и 43 %, у 66 % первичным амилоидозом к:А — 18:48 %.
Диагноз и дифференциальный диагноз. При диагностике системного амилоидоза опираются на выявление возможной связи клинических проявлений, включая поражение почек, с предшествующим заболеванием и клинические особенности, обусловленные тропностью каждого типа амилоида к определенным органам и системам. Появление у больных группы риска АА-амилоидоза (периодическая болезнь, PA, хронические воспалительные заболевания и др.), протеинурии позволяет предполагать в первую очередь амилоидоз, учитывая частоту вовлечения почек при АА-амилоидозе. Однако отсутствие протеинурии, например у больного периодической болезнью, еще не означает отсутствия амилоидных отложений в почках, поскольку не наблюдается полного соответствия между клиническими признаками и анатомическими находками амилоида. Амилоидные депозиты при гистологическом изучении обнаруживаются во многих органах (тканях), но клинические проявления зависят от преимущественно вовлеченных органов и/или систем.
В дифференциальной диагностике системного амилоидоза следует принимать во внимание, что АА-тип — более молодой: средний возраст заболевших моложе 40 лет, при AL-амилоидозе — 65 лет. При обоих типах отмечают преобладание мужчин (1,8:1). Такой клинический признак, как макроглоссия, свойствен только AL-амилоидозу и не встречается при АА-типе. Дефицит Х-фактора коагуляции, параорбиталь-ные геморрагии, синдром карпального канала также нехарактерны для АА-амилоидоза. При АА-типе реже, чем при AL-амилоидозе, наблюдается ортостатическая гипотония.
Диагноз амилоидоза должен подтверждаться морфологически. Информативным методом и при AL-, и при АА-амилоидозе является исследование биоптата стенки прямой кишки (желательно слизистого и подслизистого слоев), при котором вероятность выявления амилоида составляет 50—70 %. При проведении биопсии пораженного органа — почки, печени и др. — амилоид удается обнаружить в 90—100 %. При AL-амилоидозе применяют также биопсию десны, костного мозга и метод аспирационной биопсии подкожной жировой клетчатки из передней брюшной стенки. При Aв2M-амилоидозе обнаружить амилоид можно в осадке синовиальной жидкости.
Для выявления амилоидных масс в гистологических срезах применяют окрасочные методы (см. «Морфологическая картина»).
Важно не только обнаружить факт наличия амилоида в тканях, но и провести его типирование, что возможно также с помощью окрасочных методов, с использованием, например, щелочного гуанидина. Указанным способом лучше диагностируется АА-амилоидоз; дифференциация AL-амилоидоза не всегда надежна. Более точные результаты получают, применяя антисыворотки (поли- и моноклональные антитела) к основным белкам амилоидных фибрилл. Для установления типа амилоида проводят иммунопероксидазную реакцию с набором (панелью) этих антисывороток.
В последние годы в клинической практике применяют метод сцинтиграфии с меченным 123I сывороточным P-компонентом (SAP) для оценки in vivo распределения амилоида в организме. P-компонент содержится в небольшом количестве (5—10 %) в амилоиде всех типов; радиоактивный SAP, введенный больному амилоидозом, специфически связывается с амилоидными депозитами; он может быть визуализирован и количественно оценен на серии сцинтиграмм. Метод особенно полезен для контроля за динамикой тканевых отложений амилоида в процессе лечения.
Течение и прогноз. Течение системного амилоидоза прогрессирующее, прогноз различается в зависимости от формы амилоидоза, сроков диагностики и степени вовлечения жизненно важных органов. На основании анализа 474 случаев AL-амилоидоза средняя продолжительность жизни у больных составляет 2—3 года. Самая низкая выживаемость отмечена у больных с застойной сердечной недостаточностью (6 мес); у больных с вовлечением почек она выше — в среднем 21 мес.
По сообщению G. Banfi и соавт., наблюдавших 65 больных с AL-амилоидозом (срок наблюдения 0,5—120 мес), 12-месячная выживаемость составила 30 % у лиц с миеломной болезнью и 70 % — у лиц с первичным амилоидозом; к 24 мес кривые выживаемости сблизились: ее показатели равнялись соответственно 30 и 50 %.
A. Bohle и соавт., изучавшие долгосрочный прогноз у 225 больных амилоидозом почек (143 человека с AA- и 82 — с AL-типом), установили, что независимо от типа амилоидоза прогноз хуже, если на время биопсии (подтверждения диагноза) у больных наблюдался нефротический синдром; прогноз был значительно хуже, если отмечались эпизоды ОПН и выявлялись морфологические признаки интерстициального фиброза.
Среднюю продолжительность жизни у больных с АА-амилоидозом трудно оценить из-за гетерогенности этой популяции, но она больше, чем у больных AL-амилоидозом; по данным D. Wegelius и соавт., она составила 30—60 мес. Согласно данным Л.Н. Кочубей и соавт., основанным на большом материале клиники им. Е.М. Тареева, средняя продолжительность жизни больных составила 13,3 года при вторичном и 6,7 года — при естественном течении периодической болезни. Показатели 5-и 10-летней выживаемости в общей группе больных вторичным АА-амилоидозом от момента выявления протеинурии составили соответственно 77 и 44 %, у больных периодической болезнью — 48 и 24 %. Показатель 10-летней выживаемости (отдаленный прогноз) зависел от характера предлежащего заболевания и возможности его эффективного лечения: у больных ревматоидным артритом (болезнью Бехтерева) и хроническими воспалительными заболеваниями кишечника он был ниже, чем у больных с хроническими нагноениями и туберкулезом, и при всех этих заболеваниях — ниже у больных с активным или постоянно рецидивирующим течением, чем у больных с достигнутой ремиссией.
Лечение амилоидоза должно быть направлено на уменьшение синтеза предшественников, из которых строится белок амилоида.
Лечение АА-амилоидоза включает обязательное удаление источника продукции SAA — опухоли, костных секвестров, резекцию кишки (при тяжелых осложненных формах неспецифического язвенного колита или болезни Крона), излечение туберкулеза, других хронических инфекций. При длительном лечении ревматоидного артрита (более года) цитостатиками — циклофосфаном, хлорамбуцилом (обсуждается также эффективность лечения метотрексатом) амилоидоз возникает реже, а при уже развившемся амилоидозе наблюдается уменьшение его клинических проявлений — стабилизация почечной функции и снижение протеинурии вплоть до исчезновения нефротического синдрома, нормализация уровня С-реактивного белка и, вероятно, SAA. Так, в контролируемом исследовании, проведенном в середине 80-х годов, сравнивали 2 группы пациентов (в каждой по 11 больных АА-амилоидозом, развившимся на фоне PA), одни из которых получали цитостатики (хлорамбуцил, циклофосфамид или азатиоприн), а другие — не получали такого лечения. В первой группе только у 2 больных через 54 мес развилась ХПН, у остальных функция почек оставалась стабильной, наблюдалось либо исчезновение нефротического синдрома, либо уменьшение протеинурии на 50 %. Во второй группе больных у 7 развилась почечная недостаточность в более короткие сроки (46 мес), у остальных наблюдалось усиление протеинурии или развитие нефротического синдрома. В другом (неконтролируемом) многолетнем (10—21 год) наблюдении среди 16 больных PA, ЮРА и анкилозирующим спондилоартритом с АА-амилоидозом, которых лечили повторными курсами хлорамбуцила и/или циклофосфамида, у 75 % функция почек оставалась сохранной в течение 10 лет, у большинства больных отмечено снижение протеинурии и у всех — нормализация уровня С-реактивного белка.
При лечении АА-амилоидоза в рамках периодической болезни препаратом выбора является колхицин, который в эксперименте блокирует образование амилоидускоряющего фактора (АУФ), ингибирует синтез и секрецию SAA, влияет на хемотаксическую активность полиморфно-ядерных лейкоцитов крови. Лечение колхицином может предупредить возникновение амилоидоза у больных периодической болезнью. При уже возникшем амилоидозе почек проводят длительное (пожизненное) лечение колхицином в дозе 1,5—2 мг (при почечной недостаточности дозу уменьшают соответственно степени снижения клубочковой фильтрации). По данным О.М. Виноградовой, Л.Н. Кочубей, Т.В. Чегаевой, положительный эффект (уменьшение или полное исчезновение HC и протеинурии) наблюдался у 95 % из 47 больных периодической болезнью с амилоидозом, получавших колхицин в течение 4 лет. Препарат оказывал положительное влияние и у больных с начальной стадией ХПН (уровень креатинина крови менее 0,264 ммоль/л), вызывая стабилизацию почечной функции. Изучение выживаемости показало, что в результате применения колхицина средняя продолжительность жизни больных периодической болезнью с амилоидозом от момента выявления протеинурии возросла с 6,7 до 16 лет. Колхицин достаточно хорошо переносится, возникающие иногда диспепсические явления обычно не требуют полной отмены препарата. Имеются наблюдения успешного применения колхицина и при вторичном АА-амилоидозе.
Признается положительный эффект диметилсулъфоксида — ДМСО, обладающего прямым рассасывающим (резорбтивным) действием на амилоидные отложения. Препарат может быть эффективным лишь в дозе не менее 10 г/сут при продолжительности лечения 6 мес и более, что не всегда удается выполнить из-за нередких аллергий и неприятного запаха изо рта, связанного с особенностями метаболизма препарата. Определенный эффект может быть достигнут при лечении делагилом, особенно у больных с вторичным амилоидозом в рамках ревматоидного артрита. Применение сырой печени по-прежнему рекомендуется в качестве пищевой добавки.
При AL-амилоидозе, как и миеломной болезни, применяют различные схемы полихимиотерапии с целью уменьшения продукции легких цепей иммуноглобулинов. Наиболее хорошо проанализирована схема мелфалан — преднизолон. Применяются интермиттирующие схемы (мелфалан по 0,15 мг/кг и преднизолон по 0,8 мг/кг 7-дневными курсами с перерывом 4—6 нед), так как мелфалан является токсичным препаратом и среди отдаленных последствий его применения возможно развитие второй опухоли (острый лейкоз или другие заболевания, в частности миелодиспластический синдром). Лечение должно быть длительным — не менее 6 мес — 1 года. У пациентов с почечной недостаточностью (клиренс креатинина менее 40 мл/мин) дозу мелфалана необходимо снизить на 50 %; терапия прекращается, если через 3 мес лечения отмечается дальнейшее снижение почечной функции.
Вероятность положительного эффекта терапии у больных с HC выше при исходно нормальном уровне креатинина крови и отсутствии амилоидной кардиомиопатии. По результатам крупного исследования, включившего 153 больных с AL-амилоидозом, при лечении по схеме мелфалан — преднизолон в течение 24—36 мес положительный эффект наблюдался в среднем у 18 % больных; у больных с сердечной недостаточностью результаты хуже. Продолжительность жизни больных, ответивших на лечение, может составить 5 лет, что по сравнению с естественным течением означает большой прогресс.
В наблюдении G. Merlini и соавт. (лечение по схеме мелфалан — преднизолон не менее 6 мес оказалось успешным у 38 из 97 пациентов AL-ами-лоидозом с улучшением выживаемости (в среднем до 120 мес против 14 мес в контроле). В наблюдении G. Banfi, G. Marinone 40 больных AL-амилоидозом с поражением почек, принимавших схемы мелфалан — преднизолон по крайней мере 6 мес, были далее прослежены в течение в среднем 36 мес. Среди ответивших на лечение за этот срок только у 4 больных развилась терминальная почечная недостаточность, в то время как среди больных, не ответивших на терапию, 50 % достигли терминальной ХПН и 70 % умерли в течение 12 мес. Монотерапия колхицином при AL-амилоидозе оказалась неэффективной, добавление колхицина к схеме мелфалан — преднизолон также не привело к существенному улучшению результатов.
В последнее время у больных AL-амилоидозом моложе 60 лет все чаще применяют более агрессивные схемы полихимиотерапии с включением винкристина, доксорубицина, циклофосфана, мелфалана, дексаметазона в разных комбинациях с последующей трансплантацией стволовых клеток.
Лечение застойной сердечной недостаточности у больных с AL-амилоидозом сводится, по сути дела, только к назначению массивных доз мочегонных средств, так как отмечается повышенная чувствительность к сердечным гликозидам, ингибиторам кальциевых каналов, в-блокаторам. В настоящее время для лечения амилоидной кардиомиопатии начинает внедряться трансплантация сердца. Пока о результатах этого метода лечения судить преждевременно.
При ортостатической гипотонии полезна диета с высоким содержанием поваренной соли (более 6 г/сут), назначают также минералокор-тикоиды. При диарее показана диета с пониженным содержанием жиров, растительной клетчатки, ограничение потребления жидкости.
Современные методы трансплантационной терапии и экстракорпоральные методы вносят большие изменения в лечение амилоидной нефропатии на стадии почечной недостаточности. Больные с ХПН на гемодиализе живут примерно столько же, сколько и другие больные с системными заболеваниями, например сахарным диабетом. При проведении гемодиализа возникают осложнения, связанные с фистулой, гипотонические кризы из-за поражения надпочечников и ортостатической гипотонии. Кроме того, гемодиализ не защищает больного от прогрессирования амилоидоза, может наступить смерть в связи с амилоидозом других локализаций, например сердца, даже при вторичном амилоидозе, для которого тяжелое поражение сердца не столь характерно. Некоторые преимущества перед гемодиализом имеет амбулаторный перитонеальный диализ, поскольку при его проведении не требуется сосудистый доступ, из-за малых кровопотерь удается поддерживать достаточный уровень гематокрита у больных. Однако при перитонеальном диализе возможны инфекционные осложнения, особенно в условиях продолжения медикаментозного лечения амилоидоза, включающего цитостатики. Кроме того, описаны случаи инфильтрации амилоидом брюшины, что снижает функциональные возможности этого природного фильтра.
Хорошая или удовлетворительная реабилитация достигается более чем у 60 % больных независимо от типа амилоидоза (AA или AL). Так, в наблюдении G. Moroni и соавт. среди 43 пациентов, находящихся на хроническом гемодиализе (16 больных с AL- и 21- с АА-амилоидозом), 68 % прожили в течение года и 30 % — 5 лет от начала заместительной терапии. Выживаемость больных, находящихся на перитонеальном диализе, сходна с этим показателем у больных, получающих гемодиализ.
Трансплантация почек — более перспективный метод у больных амилоидозом AA- и AL-типа с почечной недостаточностью. Она показана больным с медленно прогрессирующим течением заболевания и отсутствием тяжелого амилоидного поражения сердца и желудочно-кишечного тракта.
После трансплантации показатели выживаемости больного и трансплантата сопоставимы с этими показателями в других группах пациентов с пересаженной почкой. В наблюдении Hartmann и соавт. показатель 3-летней выживаемости больных составил 51 %, 10-летней — 43 %; соответствующие показатели выживаемости трансплантата — 62 и 41 %. Неблагоприятными прогностическими факторами при трансплантации почек у больных амилоидозом AA- и AL-типа были возраст более 40 лет и многоорганные вовлечения. В среднем через 3 года после трансплантации возникает реамилоидоз в трансплантированной почке, что доказано как при биопсии трансплантата, так и при сцинтиграфическом исследовании. В наблюдении К.L. Harrison и соавт. среди больных АА-амилоидозом с трансплантированной почкой у лиц с периодической болезнью (17 %) и хронической инфекцией (13 %) реамилои-доз возникал чаще, чем у лиц с PA и анкилозирующим спондилоартритом (6 %); у больных с первичным AL-амилоидозом — у 28 %. Потеря трансплантата отмечена у 2 % больных с АА-амилоидозом и у 3 % — с AL-типом.
В лечении семейного ATTR-амилоидоза основным направлением является трансплантация печени — органа, в котором происходит синтез транстиретина; к 1995 г. сообщено более чем о 60 таких трансплантаций. Трансплантация печени приводит к улучшению (восстановлению) автономных (вегетативных) функций (сфинктерных нарушений, кишечных расстройств, ортостатической гипотонии). Что касается периферической нейропатии, то восстановить ее не удается, если развились необратимые дистрофические изменения нервных стволов. Кроме того, трансплантация печени оказывается менее эффективной при одновременном поражении сердца и гипотонии.
Проблема лечения диализного амилоидоза изучена мало. Наилучшие результаты удается достигнуть при трансплантации почек, в частности, быстро уменьшается до нормального уровень в2M, исчезают клинические признаки амилоидоза, однако отложения амилоида прослеживаются долго — более 10 лет.
Сохраняется значение симптоматического лечения. При наличии синдрома карпального канала используют наложение шин на запястье, тепловые процедуры и двигательные упражнения, при шейной деструктивной спондилоартропатии — воротниковую иммобилизацию шеи. По данным J. Kay, аппликации на лопатку 10 % гидрокортизоновой мази с помощью фонофореза или внутрисуставные инъекции кортикостероидов заметно уменьшают боль, вызванную плечелопаточным периартритом. Хирургическое рассечение карпальной связки может быть неэффективным, так как известны случаи повторного развития симптомов сдавления срединного нерва. Необходимость в хирургическом вмешательстве с целью повышения прочности костно-суставных структур возникает, как правило, при накоплении больших масс амилоида.







 26.01.2018
26.01.2018